Glyoxylate metabolism (WP5166)
Homo sapiens

{kind=link}
{kind=link}
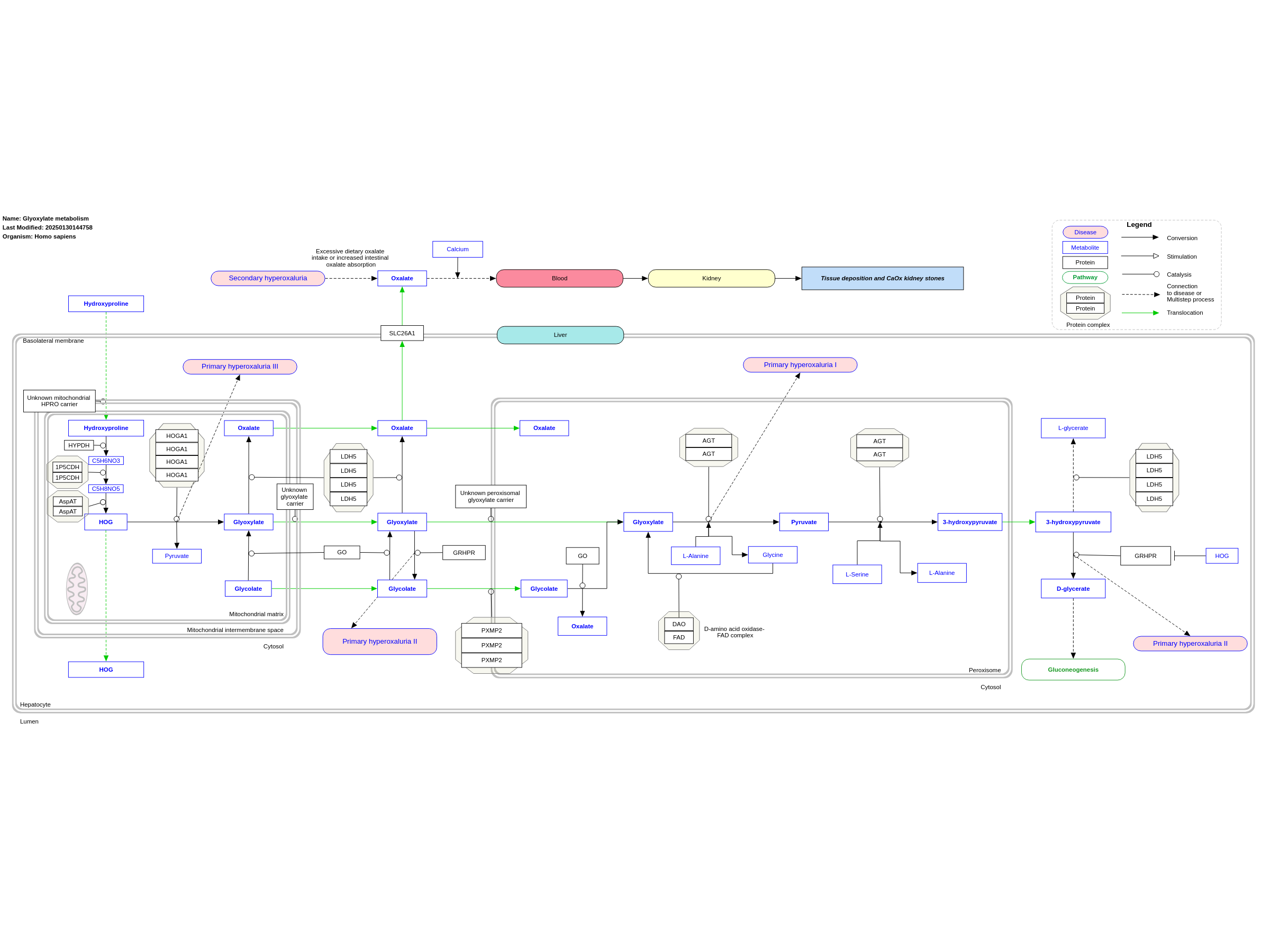
The glyoxylate metabolism in hepatocytes is affected by primary hyperoxaluria (PH) types 1-3, leading to glyoxylate accumulation and hence, increased oxalate production, which is transported out of the hepatocytes by SLC26a1 transporters on the basolateral membrane into the blood and consequently, the kidneys, where it causes the occurrence of CaOx (calcium + oxalate) crystal deposition and hence, kidney stones. PH1 is caused due to a mutation of the AGT (glyoxylate aminotransferase) trimer, responsible for the conversion of glyoxylate into pyruvate in the peroxisome. PH2 is caused by mutations of glyoxylate reductase (GR), that converts 3-hydroxypyruvate into D-glycerate in the cytosol. It also catalyses the conversion of glyoxylate into glycolate. PH3 is linked to mutations on the HOGA1 gene, yielding the tetramer 4‐hydroxy‐2‐oxoglutarate aldolase, which acts in the mitochondrion to convert 4-hydroxy-2-oxoglutarate to glyoxylate. Secondary hyperoxaluria is caused by (1) increased absorption of dietary oxalate through the GI tract or (2) increased consumption of dietary oxalate. There is an abundance of knowledge gaps in this pathway, specifically regarding the peroxisomal and mitochondrial transporters for several metabolites. This pathway is based on Physicians Guide to the Diagnosis, Treatment, and Follow-up of Inherited Metabolic Diseases by Nenad Blau Chapter 28 (Hyperoxalurias) (ISBN 3642403360).
For a description of pathway objects, see the WikiPathways Legend.
Authors
Emilia Agasi , Alexandra Bosch , Egon Willighagen , Denise Slenter , and Eric WeitzActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways ONTOX Rare DiseasesAnnotations
Pathway Ontology
D-alanine metabolic pathway disease pathway glycine metabolic pathway primary hyperoxaluria type 2 pathway hyperoxaluria pathway primary hyperoxaluria pathway primary hyperoxaluria type 1 pathwayCell Type Ontology
hepatocyte native cellDisease Ontology
primary hyperoxaluria type 2 inherited metabolic disorder primary hyperoxaluria primary hyperoxaluria type 3 primary hyperoxaluria type 1| Label | Type | Compact URI | Comment |
|---|---|---|---|
| C5H6NO3 | Metabolite | chebi:62612 | (3R,5S)-1-pyrroline-3-hydroxy-5-carboxylate |
| Calcium | Metabolite | chebi:29108 | |
| HOG | Metabolite | chebi:17742 | 4-hydroxy-2-oxoglutarate |
| Pyruvate | Metabolite | chebi:15361 | |
| Glyoxylate | Metabolite | chebi:36655 | |
| Oxalate | Metabolite | chebi:30623 | |
| Pyruvate | Metabolite | chebi:15361 | |
| L-Alanine | Metabolite | chebi:57972 | |
| Glycine | Metabolite | chebi:57305 | |
| 3-hydroxypyruvate | Metabolite | chebi:17180 | |
| L-Serine | Metabolite | chebi:33384 | |
| D-glycerate | Metabolite | chebi:16659 | |
| L-glycerate | Metabolite | wikidata:Q27102017 | |
| HOG | Metabolite | chebi:17742 | What is happening here?4-hydroxy-2-oxoglutarate |
| FAD | Metabolite | chebi:16238 | Flavin adenine dinucleotide |
| Hydroxyproline | Metabolite | chebi:18095 | trans-4-hydroxy-L-proline |
| Glycolate | Metabolite | chebi:29805 | |
| C5H8NO5 | Metabolite | chebi:6331 | erythro-4-hydroxy-L-glutamate(1−) |
| SLC26A1 | Protein | uniprot:Q9H2B4 | |
| HYPDH | Protein | uniprot:Q9UF12 | Hydroxyproline dehydrogenase |
| PXMP2 | Protein | uniprot:Q9NR77 | Peroxisomal membrane protein 2 |
| HOGA1 | Protein | uniprot:Q86XE5 | 4-hydroxy-2-oxoglutarate aldolase 1 |
| GRHPR | Protein | uniprot:Q9UBQ7 | Also called D-glycerate dehydrogenase (GDH), and hydroxypyruvate reductaseAbbreviated with GR or GRHPR |
| GO | Protein | uniprot:Q9UJM8 | |
| LDH5 | Protein | uniprot:P00338 | L-lactate dehydrogenase |
| DAO | Protein | uniprot:P14920 | D-amino-acid oxidase |
| AGT | Protein | uniprot:P21549 | Serine-pyruvate transaminase, SPTAlso: Alanine-glyoxylate aminotransferase |
| HOGA1 | Protein | uniprot:Q86XE5 | 4-hydroxy-2-oxoglutarate aldolase |
| 1P5CDH | Protein | uniprot:P30038 | Delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial |
| AspAT | Protein | uniprot:P00505 | aspartate aminotransferase |
| GO | Protein | uniprot:Q9UJM8 | Glycolate oxidaseHydroxyacid oxidase |
| LDH5 | Protein | uniprot:P00338 | L-lactate dehydrogenaseConsists of four M-subunits (found in muscle and liver), coded by LDHA gene |
| Unknown peroxisomalglyoxylate carrier | Protein | reactome:R-HSA-6784434 | Cytosolic glyoxylate can enter the peroxisome but the carrier that mediates its entry has not been identified (Wanders et al. 2016). |
| Unknownglyoxylatecarrier | Protein | reactome:R-HSA-6784436 | |
| Unknown mitochondrialHPRO carrier | Protein | reactome:R-HSA-6784213 | Id: R-HSA-8953316.2 (found on Reactome) |
| AGT | Protein | uniprot:P21549 | Serine-pyruvate transaminaseAlso: Alanine-glyoxylate aminotransferase |
References
- Carrier-mediated transport controls hydroxyproline catabolism in heart mitochondria from spontaneously hypertensive rat. Atlante A, Seccia TM, Marra E, Minervini GM, Vulpis V, Pirrelli A, et al. FEBS Lett. 1996 Nov 4;396(2–3):279–84. PubMed Europe PMC Scholia
- Pxmp2 is a channel-forming protein in Mammalian peroxisomal membrane. Rokka A, Antonenkov VD, Soininen R, Immonen HL, Pirilä PL, Bergmann U, et al. PLoS One. 2009;4(4):e5090. PubMed Europe PMC Scholia
- Metabolism of (13)C5-hydroxyproline in mouse models of Primary Hyperoxaluria and its inhibition by RNAi therapeutics targeting liver glycolate oxidase and hydroxyproline dehydrogenase. Li X, Knight J, Fargue S, Buchalski B, Guan Z, Inscho EW, et al. Biochim Biophys Acta. 2016 Feb;1862(2):233–9. PubMed Europe PMC Scholia
- Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Wanders RJA, Waterham HR, Ferdinandusse S. Front Cell Dev Biol. 2016 Jan 28;3:83. PubMed Europe PMC Scholia
- Dihydrodipicolinate Synthase: Structure, Dynamics, Function, and Evolution. Grant Pearce F, Hudson AO, Loomes K, Dobson RCJ. Subcell Biochem. 2017;83:271–89. PubMed Europe PMC Scholia
- Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Lai C, Pursell N, Gierut J, Saxena U, Zhou W, Dills M, et al. Mol Ther. 2018 Aug 1;26(8):1983–95. PubMed Europe PMC Scholia
- Downregulated Expression of Solute Carrier Family 26 Member 6 in NRK-52E Cells Attenuates Oxalate-Induced Intracellular Oxidative Stress. Jiang H, Gao X, Gong J, Yang Q, Lan R, Wang T, et al. Oxid Med Cell Longev. 2018 Oct 10;2018:1724648. PubMed Europe PMC Scholia
- Lactate dehydrogenase 5: identification of a druggable target to reduce oxaluria. Stevens JS, Al-Awqati Q. J Clin Invest. 2019 May 20;129(6):2201–4. PubMed Europe PMC Scholia
- Regulation of human 4-hydroxy-2-oxoglutarate aldolase by pyruvate and α-ketoglutarate: implications for primary hyperoxaluria type-3. Huang A, Burke J, Bunker RD, Mok YF, Griffin MD, Baker EN, et al. Biochem J. 2019 Nov 15;476(21):3369–83. PubMed Europe PMC Scholia
- Dimerization Drives Proper Folding of Human Alanine:Glyoxylate Aminotransferase But Is Dispensable for Peroxisomal Targeting. Dindo M, Ambrosini G, Oppici E, Pey AL, O’Toole PJ, Marrison JL, et al. J Pers Med. 2021 Apr 6;11(4):273. PubMed Europe PMC Scholia