Disorders of fructose metabolism (WP5178)
Homo sapiens

{kind=link}
{kind=link}
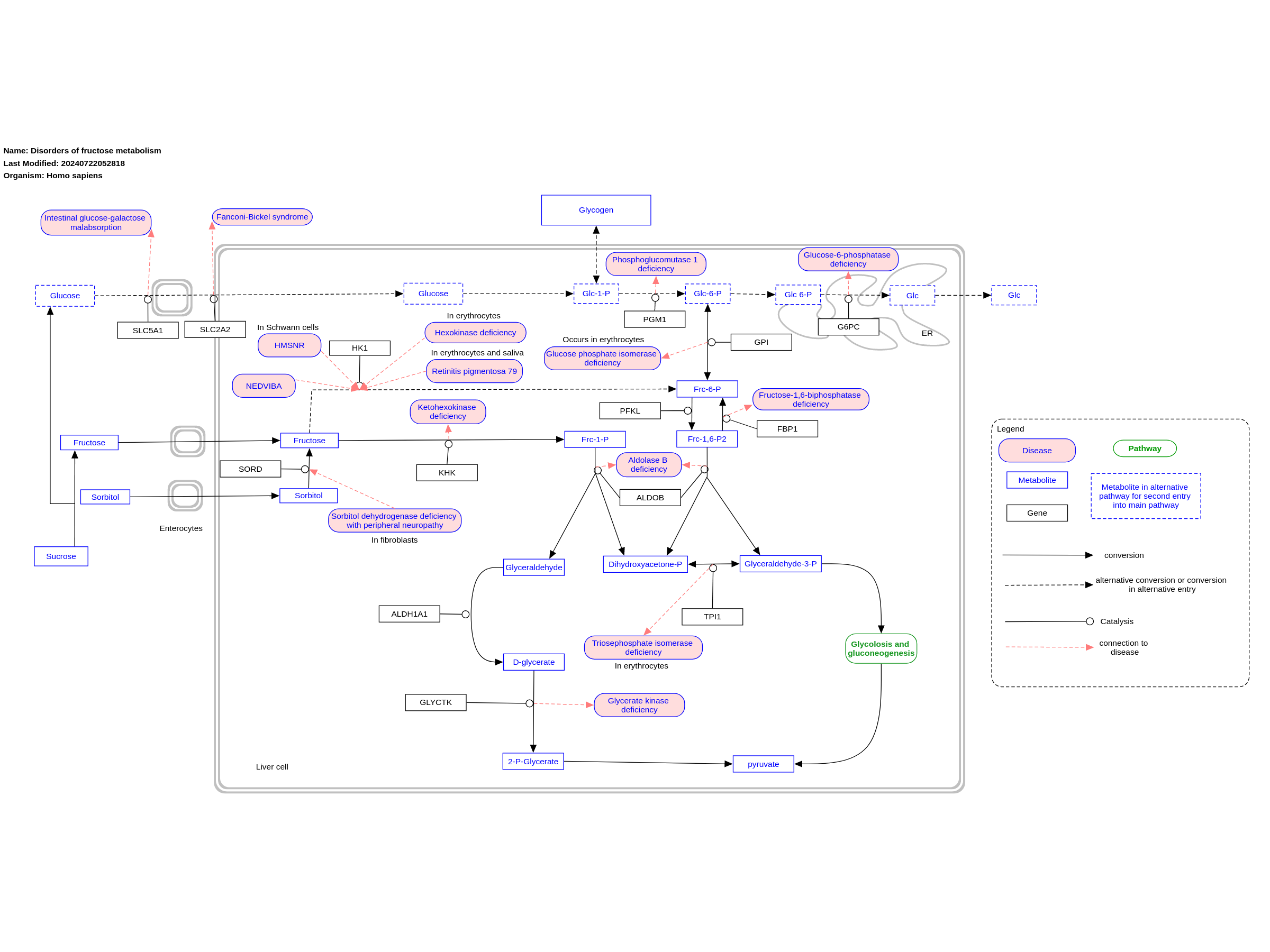
Fructose is converted into fructose-1-phosphate, this reaction is facilitated by fructokinase (also called ketohexokinase)(encoded by KHK). Connected to this reaction is the metabolic disease “essential fructosuria” which causes a deficiency in fructokinase. After this fructose-1-phosphate is converted into glyceraldehyde and dihydroxyacetone-phosphate, this is facilitated by aldolase B (encoded by ALDOB). connected to this reaction is the metabolic disease “hereditary fructose intolerance”, which is the result of the absence of aldolase B. The glyceraldehyde produced from this reaction will be converted into D-glycerate through the activity of Aldehyde dehydrogenase 1 family, member A1 or ALDH1A1 (encoded by the ALDH1A1 gene). The D-glycerate will be converted (according to the book) into 2-Phosphoglyceric acid through the activity of Glycerate kinase (encoded by GLYCTK), connected to this reaction is the metabolic disease “glycerate kinase deficiency” which results in the accumulation of D-glycerate. The resulting 2-phosphoglyceric acid then goes through a series of reactions resulting in pyruvate. Another entry into this pathway is through the conversion of glucose-6-phosphate into fructose-6-phosphate through glucose-6-phosphate isomerase (encoded by GPI). The fructose-6-phosphate can be converted into fructose-1,6-biphosphate through phosphofructokinase (encoded by PFKL). Fructose-1,6-biphosphate can be converted into fructose-6-phosphate by fructose-1,6-biphosphatase, connected to this reaction is the metabolic disease “Fructose-1,6-biphosphatase deficiency”. When going further, the fructose-1,6-biphosphate is converted into dihydroxyacetone-phosphate and glyceraldehyde-3-phosphate through the activity of aldolase B, this reaction is also affected by hereditary fructose intolerance. The glyceraldehyde-3-phosphate is then eventually turned into pyruvate through a series of reactions. The dihydroxyacetone-phosphate resulting from the aldolase B involved reactions can be converted into glyceraldehyde-3-phosphate through the activity of triose-phosphate isomerase (encoded by TPI1). This pathway is based on the fructose metabolism pathway (Chapter 18, Figure 18.5) in the book Physicians Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases (ed. 4) by Nenad Blau, Marinus Duran, K Michael Gibson, Carlo Dionisi.(ISBN 3642403360 (978-3642403361))
For a description of pathway objects, see the WikiPathways Legend.
Authors
Enzo Chiaradia , Sam Drabbe , Egon Willighagen , Lars Willighagen , and Eric WeitzActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways ONTOX Rare Diseases Serious Request 2024 - MetaKidsAnnotations
Disease Ontology
essential fructosuria congenital disorder of glycosylation D-glyceric aciduria fructose-1,6-bisphosphatase deficiency retinitis pigmentosa hereditary fructose intolerance syndromePathway Ontology
carbohydrate metabolic pathway inborn error of fructose metabolism pathway| Label | Type | Compact URI | Comment |
|---|---|---|---|
| Sorbitol | Metabolite | chebi:30911 | |
| Glc-1-P | Metabolite | chebi:16077 | |
| Glyceraldehyde-3-P | Metabolite | chebi:29052 | |
| Glyceraldehyde | Metabolite | chebi:17378 | |
| Sucrose | Metabolite | chebi:17992 | |
| Glucose | Metabolite | chebi:17234 | |
| Frc-1,6-P2 | Metabolite | chebi:32966 | |
| Frc-6-P | Metabolite | chebi:57634 | |
| Glycogen | Metabolite | chebi:28087 | |
| Fructose | Metabolite | chebi:28645 | |
| 2-P-Glycerate | Metabolite | chebi:17835 | |
| pyruvate | Metabolite | chebi:15361 | |
| D-glycerate | Metabolite | chebi:16659 | |
| Frc-1-P | Metabolite | chebi:138881 | |
| Dihydroxyacetone-P | Metabolite | chebi:16108 | |
| Glc-6-P | Metabolite | chebi:58225 | |
| Glc | Metabolite | chebi:17234 | |
| Glc 6-P | Metabolite | chebi:4170 | |
| SLC2A2 | GeneProduct | ensembl:ENSG00000163581 | |
| ALDH1A1 | GeneProduct | ensembl:ENSG00000165092 | |
| PGM1 | GeneProduct | ensembl:ENSG00000079739 | |
| GLYCTK | GeneProduct | ensembl:ENSG00000168237 | |
| PFKL | GeneProduct | ensembl:ENSG00000141959 | |
| GPI | GeneProduct | ensembl:ENSG00000105220 | |
| G6PC | GeneProduct | ensembl:ENSG00000131482 | |
| SORD | GeneProduct | ensembl:ENSG00000140263 | |
| FBP1 | GeneProduct | ensembl:ENSG00000165140 | |
| SLC5A1 | GeneProduct | ensembl:ENSG00000100170 | |
| TPI1 | GeneProduct | ensembl:ENSG00000111669 | |
| KHK | GeneProduct | ensembl:ENSG00000138030 | |
| ALDOB | GeneProduct | ensembl:ENSG00000136872 | |
| HK1 | GeneProduct | ensembl:ENSG00000156515 |
References
- Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases [Internet]. Blau N, Duran M, Gibson KM, Dionisi Vici C, editors. Springer Berlin Heidelberg; 2014. Available from: http://dx.doi.org/10.1007/978-3-642-40337-8 DOI Scholia
- Escherichia coli ☆. Neidhardt FC, Kushner SR. In: Reference Module in Life Sciences [Internet]. Elsevier; 2017. Available from: http://dx.doi.org/10.1016/B978-0-12-809633-8.06393-7 DOI Scholia
- Activity and specificity of human aldolases. Gamblin SJ, Davies GJ, Grimes JM, Jackson RM, Littlechild JA, Watson HC. J Mol Biol. 1991 Jun 20;219(4):573–6. PubMed Europe PMC Scholia
- Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Diabetes. 2003 Sep;52(9):2426–32. PubMed Europe PMC Scholia
- D-glyceric aciduria is caused by genetic deficiency of D-glycerate kinase (GLYCTK). Sass JO, Fischer K, Wang R, Christensen E, Scholl-Bürgi S, Chang R, et al. Hum Mutat. 2010 Dec;31(12):1280–5. PubMed Europe PMC Scholia
- New insights into the half-of-the-sites reactivity of human aldehyde dehydrogenase 1A1. Yoval-Sánchez B, Pardo JP, Rodríguez-Zavala JS. Proteins. 2013 Aug;81(8):1330–9. PubMed Europe PMC Scholia
- The metabolic alterations of cancer cells. Sciacovelli M, Gaude E, Hilvo M, Frezza C. Methods Enzymol. 2014;542:1–23. PubMed Europe PMC Scholia
- Two novel mutations (p.(Ser160Pro) and p.(Arg472Cys)) causing glucose-6-phosphate isomerase deficiency are associated with erythroid dysplasia and inappropriately suppressed hepcidin. Mojzikova R, Koralkova P, Holub D, Saxova Z, Pospisilova D, Prochazkova D, et al. Blood Cells Mol Dis. 2018 Mar;69:23–9. PubMed Europe PMC Scholia