Glycine metabolism, including IMDs (WP5028)
Homo sapiens

{kind=link}
{kind=link}
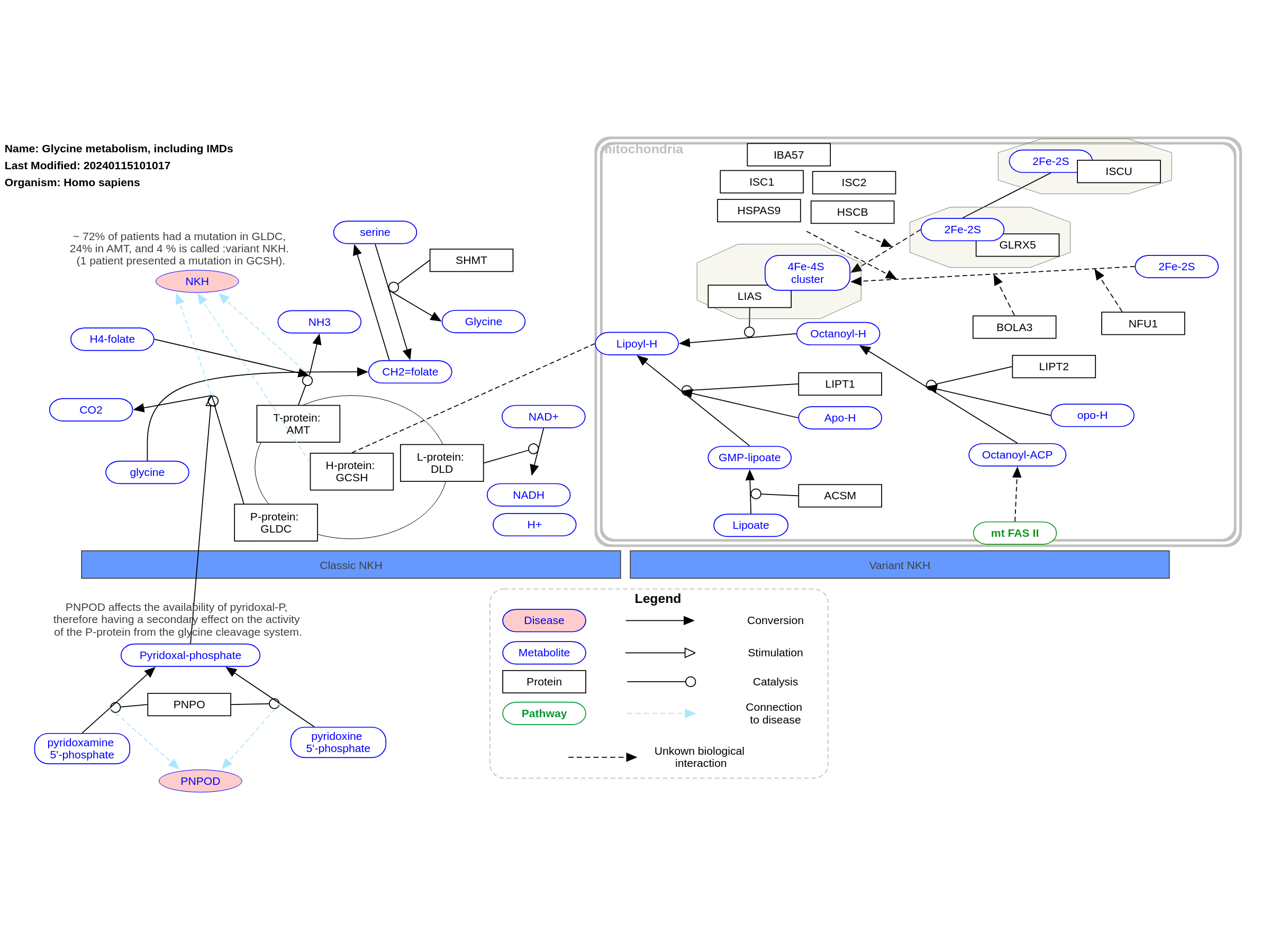
The main disorder related to glycine (NonKetotic Hyperglycinemia, NKH) is a malfunctioning of the glycine cleavage enzyme, which consists out of four subunits (P-, H-, T- and L-protein). These subunits work together (however not as a complex) to convert glycine and H4-folate into methylene-tetrahydrofolate (CH2=folate), as depicted on the lefthand side of this pathway. This disorder is also known as glycine encephalopathy, with cerebral dysfunctioning as the common denominator. Besides “classical” NKH, there are several patients without mutations in the cleavage enzyme, however presenting variants within a protein related to the formation of lipoyl-H, as depicted on the righthand side of this pathway. The individual relationship between these proteins and the formation of iron-sulfur clusters (Fe-S) are not completely known, however there are indications that mutations within the NFU1, BOLA3 and GLXR5 gene can lead to a similar phenotype as NKH; most patients present with either less or more severe neurological symptoms compared to “classical” NKH. For clarity, the influence of pyridoxal-P has been added to this pathway, where a variant within the PNPO gene can lead to secondary effects on the activity of the P-protein from the cleavage system. This pathway was inspired by Chapter 5 (edition 4) of the book of Blau (ISBN 3642403360 (978-3642403361)), Fig. 5.1.
For a description of pathway objects, see the WikiPathways Legend.
Authors
Denise Slenter , Egon Willighagen , Andra Waagmeester , Eric Weitz , Finterly Hu , and Friederike EhrhartActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways Rare DiseasesAnnotations
Disease Ontology
pyridoxamine 5'-phosphate oxidase deficiency glycine encephalopathyPathway Ontology
glycine biosynthetic pathway| Label | Type | Compact URI | Comment |
|---|---|---|---|
| H4-folate | Metabolite | chebi:137944 | AKA Tetrahydrofolic acid (THFA), or tetrahydrofolate |
| Pyridoxal-phosphate | Metabolite | chebi:597326 | |
| NAD+ | Metabolite | chebi:15846 | |
| 2Fe-2S | Metabolite | chebi:49601 | |
| 4Fe-4Scluster | Metabolite | chebi:33722 | Cofactor for mitochondrial lipoyl synthase through LIAS [https://www.uniprot.org/uniprot/O43766] |
| serine | Metabolite | chebi:17115 | |
| glycine | Metabolite | chebi:15428 | |
| GMP-lipoate | Metabolite | chebi:86459 | |
| pyridoxamine 5'-phosphate | Metabolite | chebi:58451 | |
| CO₂ | Metabolite | chebi:16526 | |
| NH3 | Metabolite | chebi:16134 | |
| Lipoate | Metabolite | chebi:30314 | |
| CH2=folate | Metabolite | chebi:1989 | aka methylene-tetrahydrofolate, 5,10-Methylenetetrahydrofolate (annotated with naturally occuring diastereoisomer ID, named [6R]-5,10-methylene-THF.). |
| Glycine | Metabolite | chebi:15428 | |
| NADH | Metabolite | chebi:16908 | |
| H⁺ | Metabolite | chebi:15378 | |
| pyridoxine 5'-phosphate | Metabolite | chebi:58589 | |
| LIPT1 | Protein | uniprot:Q9Y234 | |
| SHMT | Protein | uniprot:P34896 | Annotated with Cytosolic ID, another form is known to be active in mitochondria. |
| NFU1 | Protein | uniprot:Q9UMS0 | |
| LIAS | Protein | uniprot:O43766 | |
| GLRX5 | Protein | uniprot:Q86SX6 | |
| P-protein:GLDC | Protein | uniprot:P23378 | |
| IBA57 | Protein | uniprot:Q5T440 | |
| BOLA3 | Protein | uniprot:Q53S33 | |
| PNPO | Protein | uniprot:Q9NVS9 | |
| H-protein:GCSH | Protein | uniprot:P23434 | 'The H-protein is responsible for interacting with the three other proteins and acts as a shuttle for some of the intermediate products in glycine decarboxylation.' [https://en.wikipedia.org/wiki/Glycine_cleavage_system]After removing CO₂ from glycine, the remaining amino-methyl group ir transferred to lipoate on the H-protein |
| T-protein:AMT | Protein | uniprot:P48728 | aka GCST |
| L-protein:DLD | Protein | uniprot:P09622 | aka GCSLreduced lipoate is re-oxidized by the L-protein |
| LIPT2 | Protein | uniprot:A6NK58 | |
| ISCU | Protein | uniprot:Q9H1K1 | |
| HSCB | Protein | uniprot:Q8IWL3 |
References
- Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases [Internet]. Blau N, Duran M, Gibson KM, Dionisi-Vici C. Springer; 2014. 0 p. Available from: https://books.google.com/books/about/Physician_s_Guide_to_the_Diagnosis_Treat.html?hl=&id=wJRBnwEACAAJ OpenLibrary Worldcat
- The glycine cleavage system: structure of a cDNA encoding human H-protein, and partial characterization of its gene in patients with hyperglycinemias. Koyata H, Hiraga K. Am J Hum Genet. 1991 Feb;48(2):351–61. PubMed Europe PMC Scholia
- Defective glycine cleavage system in nonketotic hyperglycinemia. Occurrence of a less active glycine decarboxylase and an abnormal aminomethyl carrier protein. Hiraga K, Kochi H, Hayasaka K, Kikuchi G, Nyhan WL. J Clin Invest. 1981 Aug;68(2):525–34. PubMed Europe PMC Scholia
- The glycine decarboxylase system: a fascinating complex. Douce R, Bourguignon J, Neuburger M, Rébeillé F. Trends Plant Sci. 2001 Apr;6(4):167–76. PubMed Europe PMC Scholia
- Structure and properties of recombinant human pyridoxine 5’-phosphate oxidase. Musayev FN, Di Salvo ML, Ko TP, Schirch V, Safo MK. Protein Sci. 2003 Jul;12(7):1455–63. PubMed Europe PMC Scholia
- Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Kure S, Kato K, Dinopoulos A, Gail C, DeGrauw TJ, Christodoulou J, et al. Hum Mutat. 2006 Apr;27(4):343–52. PubMed Europe PMC Scholia
- Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, et al. Am J Hum Genet. 2011 Oct 7;89(4):486–95. PubMed Europe PMC Scholia
- A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, et al. Am J Hum Genet. 2011 Nov 11;89(5):656–67. PubMed Europe PMC Scholia
- Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Mayr JA, Zimmermann FA, Fauth C, Bergheim C, Meierhofer D, Radmayr D, et al. Am J Hum Genet. 2011 Dec 9;89(6):792–7. PubMed Europe PMC Scholia
- Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Baker PR 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, et al. Brain. 2014 Feb;137(Pt 2):366–79. PubMed Europe PMC Scholia
- Altering the Mitochondrial Fatty Acid Synthesis (mtFASII) Pathway Modulates Cellular Metabolic States and Bioactive Lipid Profiles as Revealed by Metabolomic Profiling. Clay HB, Parl AK, Mitchell SL, Singh L, Bell LN, Murdock DG. PLoS One. 2016 Mar 10;11(3):e0151171. PubMed Europe PMC Scholia
- Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron-sulfur proteins. Uzarska MA, Nasta V, Weiler BD, Spantgar F, Ciofi-Baffoni S, Saviello MR, et al. Elife. 2016 Aug 17;5:e16673. PubMed Europe PMC Scholia