Glycosylation and related congenital defects (WP4521)
Homo sapiens

{kind=link}
{kind=link}
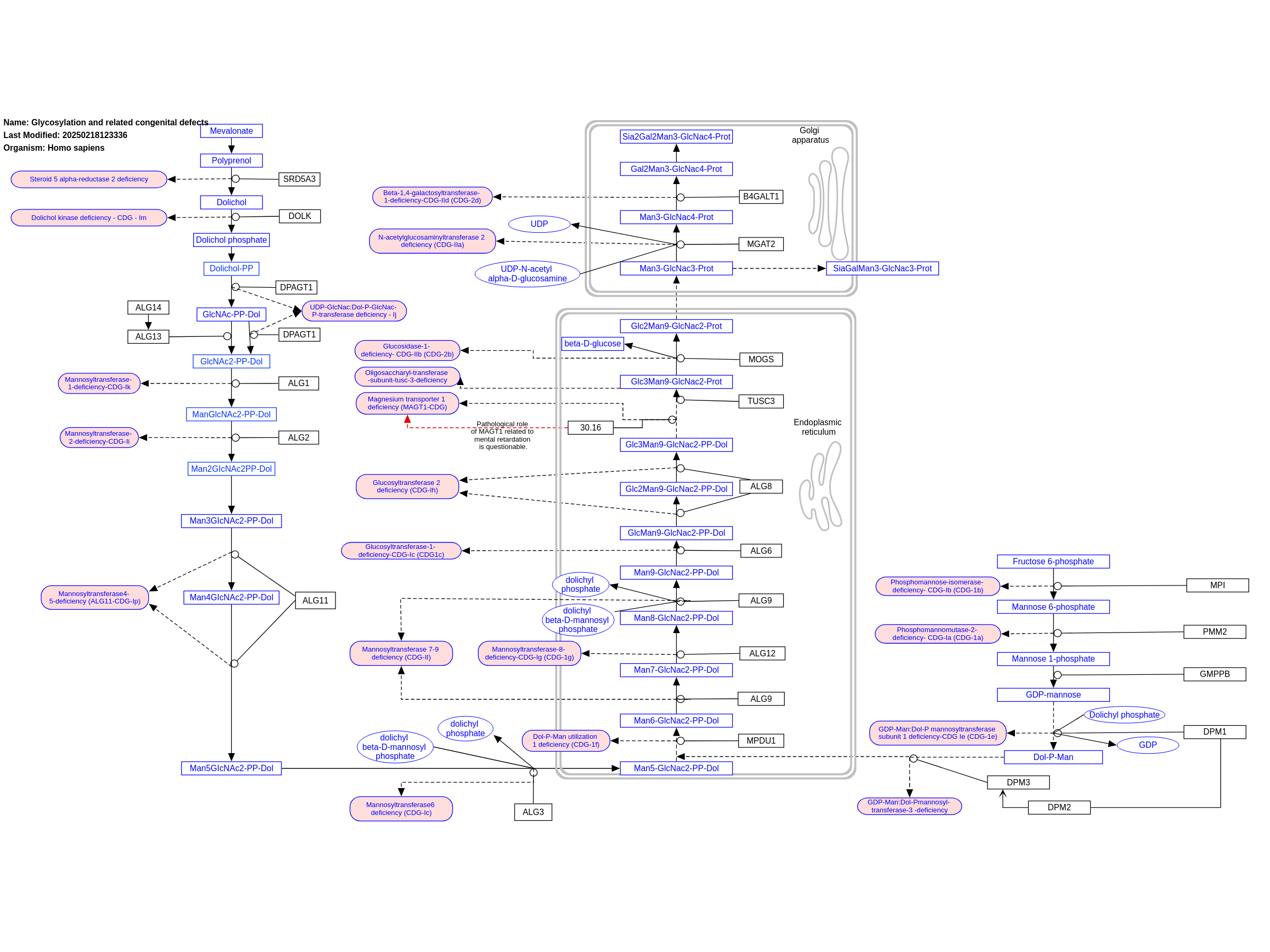
Glycosylation is the most common postranslational modification. Defects in this pathway lead to autosomal recessive disorders, called congenital disorders of glycosylation (CDG). Up to date about 50 CDGs have been identified and more are expected to be discovered. This category of metabolic disorders can be divided into four basic groups depending on where the glycosylation process occurs on the molecule. The depicts pathway depicts the group of N-glycosylation. Generally, N-glycosylation processes spread over three cellular compartments - cytosol, endoplasmic reticulum and Golgi apparatus. The associated mortality rate in combination with the limited treatment options for CDG, points out the relevance for further investigations of this pathway. Disorders resulting from an enzyme defect are highlighted in pink. This pathway was inspired by Chapter 30 of the book of Blau (ISBN 3642403360 (978-3642403361)).
For a description of pathway objects, see the WikiPathways Legend.
Authors
Eveline Schoenmaker , Denise Slenter , Britt Pieters , Lauren J. Dupuis , Irene Hemel , Egon Willighagen , Friederike Ehrhart , Finterly Hu , and Eric WeitzActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
- DNA methylation of ARHGAP30 is negatively associated with ARHGAP30 expression in lung adenocarcinoma, which reduces tumor immunity and is detrimental to patient survival (2021).
- Human Monocytes Exposed to SARS-CoV-2 Display Features of Innate Immune Memory Producing High Levels of CXCL10 upon Restimulation (2023).
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways Rare DiseasesAnnotations
Pathway Ontology
disease pathway N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminic acid dissimilation pathway altered carbohydrate metabolic pathwayDisease Ontology
autosomal recessive disease congenital disorder of glycosylation inherited metabolic disorder| Label | Type | Compact URI | Comment |
|---|---|---|---|
| dolichyl phosphate | Metabolite | chebi:57683 | |
| UDP | Metabolite | chebi:58223 | |
| beta-D-glucose | Metabolite | chebi:15903 | |
| UDP-N-acetylalpha-D-glucosamine | Metabolite | chebi:57705 | |
| GlcNAc-PP-Dol | Metabolite | chebi:58427 | In RHEA: N-acetyl-α-D-glucosaminyl-diphosphodolichol |
| dolichyl beta-D-mannosyl phosphate | Metabolite | chebi:58211 | |
| Dolichyl phosphate | Metabolite | chebi:57683 | |
| GDP | Metabolite | chebi:58189 | |
| Man5-GlcNac2-PP-Dol | Metabolite | chebi:132516 | |
| Man8-GlcNac2-PP-Dol | Metabolite | chebi:132519 | |
| Man7-GlcNac2-PP-Dol | Metabolite | chebi:132517 | |
| Man9-GlcNac2-PP-Dol | Metabolite | chebi:132520 | |
| Glc2Man9-GlcNac2-PP-Dol | Metabolite | chebi:132522 | |
| GlcMan9-GlcNac2-PP-Dol | Metabolite | chebi:132521 | |
| Glc2Man9-GlcNac2-Prot | Metabolite | chebi:59082 | |
| Glc3Man9-GlcNac2-Prot | Metabolite | chebi:132537 | |
| Man3-GlcNac4-Prot | Metabolite | chebi:60615 | |
| Man3-GlcNac3-Prot | Metabolite | chebi:60615 | |
| GDP-mannose | Metabolite | chebi:57527 | |
| Dol-P-Man | Metabolite | chebi:58211 | |
| Mannose 6-phosphate | Metabolite | chebi:58735 | |
| Mannose 1-phosphate | Metabolite | chebi:58409 | |
| Fructose 6-phosphate | Metabolite | chebi:61527 | |
| Man5GIcNAc2-PP-Dol | Metabolite | chebi:132515 | |
| Dolichol phosphate | Metabolite | chebi:57683 | In RHEA stated as dolichyl phosphate, and in this reaction it results in a -2 charge. |
| Dolichol-PP | Metabolite | chebi:15750 | In CheBi known as Dolichol diphosphate |
| Man3GIcNAc2-PP-Dol | Metabolite | chebi:132511 | |
| Dolichol | Metabolite | chebi:16091 | Full name in RHEA: di-trans,poly-cis-dolichol |
| Polyprenol | Metabolite | chebi:67132 | In RHEA and UniProt known as di-trans,cis-polyprenol. |
| Man2GIcNAc2PP-Dol | Metabolite | chebi:132510 | |
| Mevalonate | Metabolite | chebi:17710 | |
| ManGlcNAc2-PP-Dol | Metabolite | chebi:58472 | |
| GlcNAc2-PP-Dol | Metabolite | chebi:57269 | Aka N-acetylglucosamine |
| DOLK | Protein | uniprot:Q9UPQ8 | Dolichol kinase, Book label: 30.38 |
| ALG13 | Protein | uniprot:Q9NP73 | Labelled in UniProt as Putative bifunctional UDP-N-acetylglucosamine transferase and deubiquitinase ALG13.Found manually through RHEA identifier. This enzyme was not indicated in the Blau Book, but it catalyzes this exact reaction. |
| DPAGT1 | Protein | uniprot:Q9H3H5 | In UniProt described as UDP-N-acetylglucosamine--dolichyl-phosphate N-acetylglucosaminephosphotransferase.Found manually through RHEA identifier reaction. Not indicated in the Blau Book Chapter 30. |
| GMPPB | Protein | uniprot:Q9Y5P6 | AKA mannose-1-phosphate guanylyltransferase [https://en.wikipedia.org/wiki/Mannose-1-phosphate_guanylyltransferase] |
| B4GALT1 | Protein | uniprot:P15291 | AKA B4GALT1; beta-1,4-galactosyltransferase 1; book label: 30.34 |
| DPM2 | Protein | uniprot:O94777 | |
| MGAT2 | Protein | uniprot:Q10469 | AKA MGAT2; N-acetylglucosaminyltransferase 2; book label: 30.13Function'Plays an essential role in protein N-glycosylation. Catalyzes the transfer of N-acetylglucosamine (GlcNAc) onto the free terminal mannose moiety in the core structure of the nascent N-linked glycan chain, giving rise to the second branch in complex glycans.' [https://www.uniprot.org/uniprot/Q10469] |
| MOGS | Protein | uniprot:Q13724 | AKA GCS1; Glucosidase 1; book label: 30.14Function:'Cleaves the distal alpha 1,2-linked glucose residue from the Glc3Man9GlcNAc2 oligosaccharide precursor in a highly specific manner.' [https://www.uniprot.org/uniprot/Q13724] |
| TUSC3 | Protein | uniprot:Q13454 | AKA TUSC3; oligosaccharyltransferase subunit tusc 3; book label: 30.15 |
| 30.16 | Protein | uniprot:Q9H0U3 | Aka MAGT1; magnesium transporter 1 |
| ALG8 | Protein | uniprot:Q9BVK2 | AKA ALG8; Glucosyltransferase 2; book label: 30.6 |
| ALG6 | Protein | uniprot:Q9Y672 | Aka ALG6; glucosyltransferase 1; book label: 30.3 |
| ALG9 | Protein | uniprot:Q9H6U8 | book label: 30.10 |
| ALG12 | Protein | uniprot:Q9BV10 | AKA ALG12, Mannosyltransferase 8; book label: 30.5 |
| ALG9 | Protein | uniprot:Q9H6U8 | Aka ;mannosyltransferase 7-9; Book label: 30.10 |
| MPDU1 | Protein | uniprot:O75352 | AKA MPDU1; Dol-P-Man utilization 1; Book label: 30.33 |
| ALG3 | Protein | uniprot:Q92685 | AKA ALG3, mannosyltransferase 6; Book label: 30.4Function:'Adds the first Dol-P-Man derived mannose in an alpha-1,3 linkage to Man5GlcNAc2-PP-Dol' [https://www.uniprot.org/uniprot/Q92685] |
| MPI | Protein | uniprot:P34949 | AKA MPI, phosphomannose isomerase; Book label: 30.2 |
| PMM2 | Protein | uniprot:O15305 | AKA PMM, phosphomannomutase 2; book label: 30.1 |
| DPM1 | Protein | uniprot:O60762 | AKA DPM1, GDP-Man:Dol-P mannosyltransferase subunit 1; book label: 30.31 |
| DPM3 | Protein | uniprot:Q9P2X0 | AKA DPM3, GDP-Man:Dol-P mannosyltransferase 3; book label: 30.32'Stabilizer subunit of the dolichol-phosphate mannose (DPM) synthase complex; tethers catalytic subunit DPM1 to the ER' [https://www.uniprot.org/uniprot/Q9P2X0] |
| SRD5A3 | Protein | uniprot:Q9H8P0 | Known as polyprenol reductase, which catalyzes reduction of the alpha-isoprene subunit of polyprenol. But also known to have steroid dehydrogenase activity. Book label:30.39 |
| ALG1 | Protein | uniprot:Q9BT22 | Chitobiosyldiphosphodolichol beta-mannosyltransferase mediates the hexosyl group transfer.In Blau Book known as Mannosyltransferase 1, label: 30.9 |
| DPAGT1 | Protein | uniprot:Q9H3H5 | Also known as UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase, which transfers the hexosyl group. Book label: 30.8 |
| ALG11 | Protein | uniprot:Q2TAA5 | GDP-Man:Man(3)GlcNAc(2)-PP- Dol alpha-1,2-mannosyltransferase; Book label: 30.11 |
| ALG2 | Protein | uniprot:Q9H553 | Alpha-1,3/1,6- mannosyltransferase ALG2; Book label: 30.7 |
| ALG14 | Protein | ncbigene:199857 | ALG14 |
References
- Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases [Internet]. Blau N, Duran M, Gibson KM, Dionisi-Vici C. Springer; 2014. 0 p. Available from: https://books.google.com/books/about/Physician_s_Guide_to_the_Diagnosis_Treat.html?hl=&id=wJRBnwEACAAJ OpenLibrary Worldcat
- Pitfalls in early diagnosis of oral epidermoid carcinoma. Dick HM, Strelioff MG, McMurchy KA. J Can Dent Assoc (Tor). 1970 Apr;36(4):151–4. PubMed Europe PMC Scholia
- Glycosyltransferases involved in elongation of N-glycosidically linked oligosaccharides of the complex or N-acetyllactosamine type. Schachter H, Narasimhan S, Gleeson P, Vella G. Methods Enzymol. 1983;98:98–134. PubMed Europe PMC Scholia
- The human UDP-N-acetylglucosamine: alpha-6-D-mannoside-beta-1,2- N-acetylglucosaminyltransferase II gene (MGAT2). Cloning of genomic DNA, localization to chromosome 14q21, expression in insect cells and purification of the recombinant protein. Tan J, D’Agostaro AF, Bendiak B, Reck F, Sarkar M, Squire JA, et al. Eur J Biochem. 1995 Jul 15;231(2):317–28. PubMed Europe PMC Scholia
- New phenotype of mutations deficient in glucosylation of the lipid-linked oligosaccharide: cloning of the ALG8 locus. Stagljar I, te Heesen S, Aebi M. Proc Natl Acad Sci U S A. 1994 Jun 21;91(13):5977–81. PubMed Europe PMC Scholia
- Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Tan J, Dunn J, Jaeken J, Schachter H. Am J Hum Genet. 1996 Oct;59(4):810–7. PubMed Europe PMC Scholia
- Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, et al. J Clin Invest. 1998 Apr 1;101(7):1414–20. PubMed Europe PMC Scholia
- Phosphomannomutase deficiency: the molecular basis of the classical Jaeken syndrome (CDGS type Ia). Matthijs G, Schollen E, Heykants L, Grünewald S. Mol Genet Metab. 1999 Oct;68(2):220–6. PubMed Europe PMC Scholia
- Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. Körner C, Knauer R, Stephani U, Marquardt T, Lehle L, von Figura K. EMBO J. 1999 Dec 1;18(23):6816–22. PubMed Europe PMC Scholia
- A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency. De Praeter CM, Gerwig GJ, Bause E, Nuytinck LK, Vliegenthart JF, Breuer W, et al. Am J Hum Genet. 2000 Jun;66(6):1744–56. PubMed Europe PMC Scholia
- Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. Maeda Y, Tanaka S, Hino J, Kangawa K, Kinoshita T. EMBO J. 2000 Jun 1;19(11):2475–82. PubMed Europe PMC Scholia
- Biochemical and pharmacogenetic dissection of human steroid 5 alpha-reductase type II. Makridakis NM, di Salle E, Reichardt JK. Pharmacogenetics. 2000 Jul;10(5):407–13. PubMed Europe PMC Scholia
- Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. Watanabe R, Murakami Y, Marmor MD, Inoue N, Maeda Y, Hino J, et al. EMBO J. 2000 Aug 15;19(16):4402–11. PubMed Europe PMC Scholia
- Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib). Schollen E, Dorland L, de Koning TJ, Van Diggelen OP, Huijmans JG, Marquardt T, et al. Hum Mutat. 2000 Sep;16(3):247–52. PubMed Europe PMC Scholia
- Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, et al. Hum Mutat. 2000 Nov;16(5):386–94. PubMed Europe PMC Scholia
- Congenital disorders of glycosylation IIa cause growth retardation, mental retardation, and facial dysmorphism. Cormier-Daire V, Amiel J, Vuillaumier-Barrot S, Tan J, Durand G, Munnich A, et al. J Med Genet. 2000 Nov;37(11):875–7. PubMed Europe PMC Scholia
- A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If). Kranz C, Denecke J, Lehrman MA, Ray S, Kienz P, Kreissel G, et al. J Clin Invest. 2001 Dec;108(11):1613–9. PubMed Europe PMC Scholia
- Deficiency of UDP-galactose:N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. Hansske B, Thiel C, Lübke T, Hasilik M, Höning S, Peters V, et al. J Clin Invest. 2002 Mar;109(6):725–33. PubMed Europe PMC Scholia
- ALG12 mannosyltransferase defect in congenital disorder of glycosylation type lg. Grubenmann CE, Frank CG, Kjaergaard S, Berger EG, Aebi M, Hennet T. Hum Mol Genet. 2002 Sep 15;11(19):2331–9. PubMed Europe PMC Scholia
- A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation. Chantret I, Dancourt J, Dupré T, Delenda C, Bucher S, Vuillaumier-Barrot S, et al. J Biol Chem. 2003 Mar 14;278(11):9962–71. PubMed Europe PMC Scholia
- A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. Thiel C, Schwarz M, Peng J, Grzmil M, Hasilik M, Braulke T, et al. J Biol Chem. 2003 Jun 20;278(25):22498–505. PubMed Europe PMC Scholia
- Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Wu X, Rush JS, Karaoglu D, Krasnewich D, Lubinsky MS, Waechter CJ, et al. Hum Mutat. 2003 Aug;22(2):144–50. PubMed Europe PMC Scholia
- Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Kelleher DJ, Karaoglu D, Mandon EC, Gilmore R. Mol Cell. 2003 Jul;12(1):101–11. PubMed Europe PMC Scholia
- Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Schwarz M, Thiel C, Lübbehusen J, Dorland B, de Koning T, von Figura K, et al. Am J Hum Genet. 2004 Mar;74(3):472–81. PubMed Europe PMC Scholia
- Congenital disorder of glycosylation type Ik (CDG-Ik): a defect of mannosyltransferase I. Kranz C, Denecke J, Lehle L, Sohlbach K, Jeske S, Meinhardt F, et al. Am J Hum Genet. 2004 Mar;74(3):545–51. PubMed Europe PMC Scholia
- Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL. Frank CG, Grubenmann CE, Eyaid W, Berger EG, Aebi M, Hennet T. Am J Hum Genet. 2004 Jul;75(1):146–50. PubMed Europe PMC Scholia
- Accumulation of dolichol in older tissues satisfies the proposed criteria to be qualified a biomarker of aging. Parentini I, Cavallini G, Donati A, Gori Z, Bergamini E. J Gerontol A Biol Sci Med Sci. 2005 Jan;60(1):39–43. PubMed Europe PMC Scholia
- CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Weinstein M, Schollen E, Matthijs G, Neupert C, Hennet T, Grubenmann CE, et al. Am J Med Genet A. 2005 Jul 15;136(2):194–7. PubMed Europe PMC Scholia
- ALG9 mannosyltransferase is involved in two different steps of lipid-linked oligosaccharide biosynthesis. Frank CG, Aebi M. Glycobiology. 2005 Nov;15(11):1156–63. PubMed Europe PMC Scholia
- Alg14 recruits Alg13 to the cytoplasmic face of the endoplasmic reticulum to form a novel bipartite UDP-N-acetylglucosamine transferase required for the second step of N-linked glycosylation. Gao XD, Tachikawa H, Sato T, Jigami Y, Dean N. J Biol Chem. 2005 Oct 28;280(43):36254–62. PubMed Europe PMC Scholia
- In vitro evidence for the dual function of Alg2 and Alg11: essential mannosyltransferases in N-linked glycoprotein biosynthesis. O’Reilly MK, Zhang G, Imperiali B. Biochemistry. 2006 Aug 8;45(31):9593–603. PubMed Europe PMC Scholia
- Congenital disorder of glycosylation type Ia: searching for the origin of common mutations in PMM2. Quelhas D, Quental R, Vilarinho L, Amorim A, Azevedo L. Ann Hum Genet. 2007 May;71(Pt 3):348–53. PubMed Europe PMC Scholia
- A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. Kranz C, Jungeblut C, Denecke J, Erlekotte A, Sohlbach C, Debus V, et al. Am J Hum Genet. 2007 Mar;80(3):433–40. PubMed Europe PMC Scholia
- Novel 5 alpha-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Uemura M, Tamura K, Chung S, Honma S, Okuyama A, Nakamura Y, et al. Cancer Sci. 2008 Jan;99(1):81–6. PubMed Europe PMC Scholia
- A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Garshasbi M, Hadavi V, Habibi H, Kahrizi K, Kariminejad R, Behjati F, et al. Am J Hum Genet. 2008 May;82(5):1158–64. PubMed Europe PMC Scholia
- Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Molinari F, Foulquier F, Tarpey PS, Morelle W, Boissel S, Teague J, et al. Am J Hum Genet. 2008 May;82(5):1150–7. PubMed Europe PMC Scholia
- Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Lefeber DJ, Schönberger J, Morava E, Guillard M, Huyben KM, Verrijp K, et al. Am J Hum Genet. 2009 Jul;85(1):76–86. PubMed Europe PMC Scholia
- A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation-Ip. Rind N, Schmeiser V, Thiel C, Absmanner B, Lübbehusen J, Hocks J, et al. Hum Mol Genet. 2010 Apr 15;19(8):1413–24. PubMed Europe PMC Scholia
- SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, et al. Cell. 2010 Jul 23;142(2):203–17. PubMed Europe PMC Scholia
- Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic). Miller BS, Freeze HH, Hoffmann GF, Sarafoglou K. Mol Genet Metab. 2011 May;103(1):101–3. PubMed Europe PMC Scholia
- Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): extending the clinical and molecular spectrum of a rare disease. Würde AE, Reunert J, Rust S, Hertzberg C, Haverkämper S, Nürnberg G, et al. Mol Genet Metab. 2012 Apr;105(4):634–41. PubMed Europe PMC Scholia
- XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Piton A, Redin C, Mandel JL. Am J Hum Genet. 2013 Aug 8;93(2):368–83. PubMed Europe PMC Scholia
- Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Mohorko E, Owen RL, Malojčić G, Brozzo MS, Aebi M, Glockshuber R. Structure. 2014 Apr 8;22(4):590–601. PubMed Europe PMC Scholia
- Genetic defects in dolichol metabolism. Buczkowska A, Swiezewska E, Lefeber DJ. J Inherit Metab Dis. 2015 Jan;38(1):157–69. PubMed Europe PMC Scholia
- Dolichol kinase deficiency (DOLK-CDG): Two new cases and expansion of phenotype. Rush ET, Baker CV, Rizzo WB. Am J Med Genet A. 2017 Sep;173(9):2428–34. PubMed Europe PMC Scholia
- The history and recent advances in research of polyprenol and its derivatives. Sagami H, Swiezewska E, Shidoji Y. Biosci Biotechnol Biochem. 2018 Jun;82(6):947–55. PubMed Europe PMC Scholia
- GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K, Kaeser B, Ichikawa S, et al. Nat Struct Mol Biol. 2018 Mar;25(3):217–24. PubMed Europe PMC Scholia
- Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Kadirvelraj R, Yang JY, Sanders JH, Liu L, Ramiah A, Prabhakar PK, et al. Proc Natl Acad Sci U S A. 2018 May 1;115(18):4637–42. PubMed Europe PMC Scholia
- The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. Rodríguez Cruz PM, Palace J, Beeson D. Int J Mol Sci. 2018 Jun 5;19(6):1677. PubMed Europe PMC Scholia