Gamma-glutamyl cycle for the biosynthesis and degradation of glutathione, including diseases (WP4518)
Homo sapiens

{kind=link}
{kind=link}
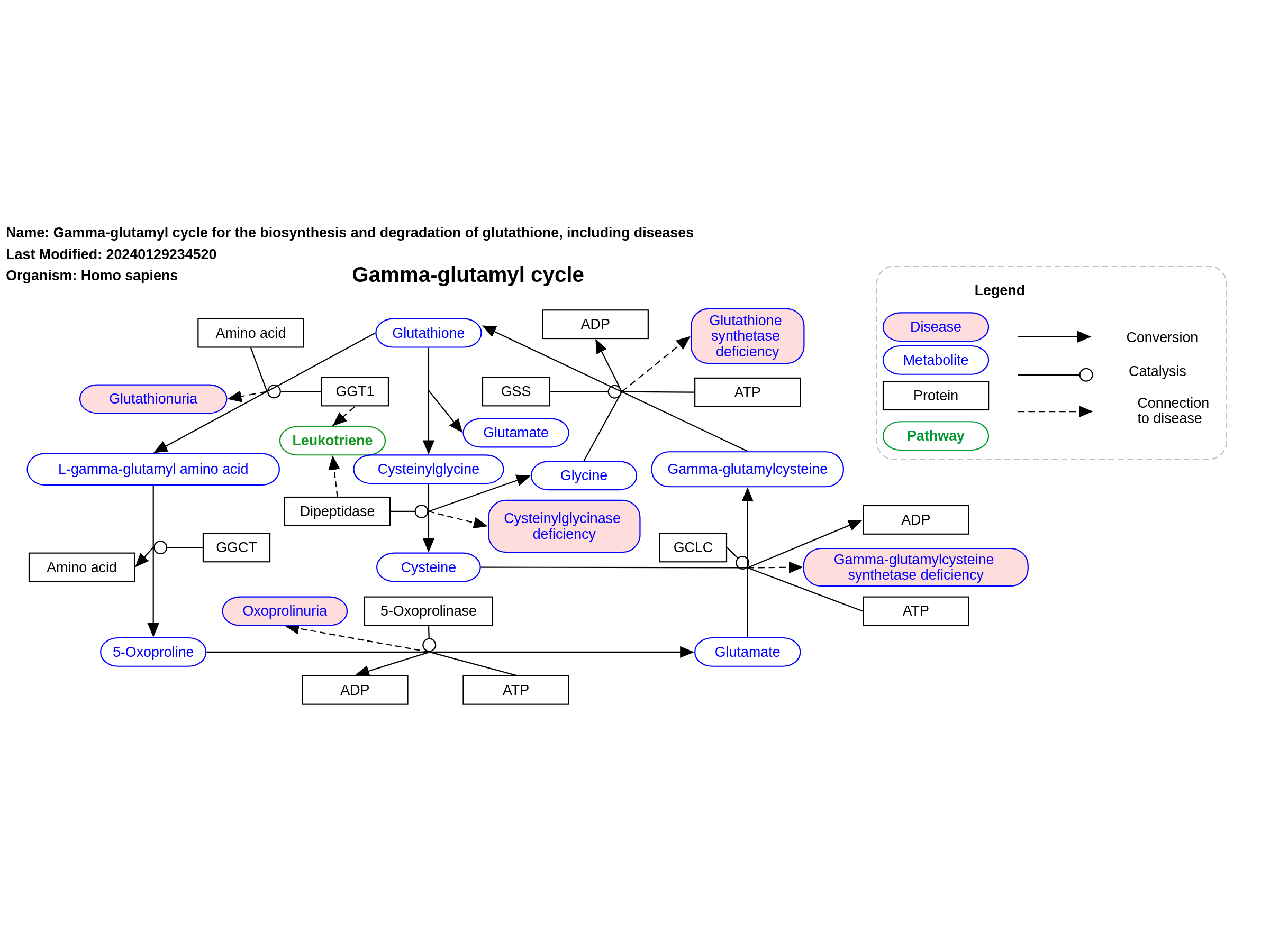
This pathway shows diseases related to the biosynthesis and degradation of glutathione. Diseases resulting from an enzyme deficiency are highlighted in pink. The four genetic defects, causing the diseases, are all inherited as autosomal recessive traits. All patients with gamma-glutamylcysteine synthetase deficiency are diagnosed with hemolytic anemia. Glutathione synthetase deficiency is classified in mild, moderate and severe. Patient diagnosed with mild glutathione synthetase deficiency suffer from hemolytic anemia only, while patient with the moderate and severe form show neurological symptoms, metabolic acidosis and bacterial infections as well. This pathway was inspired by Chapter 42 of the book of Blau (ISBN 3642403360 (978-3642403361)).
For a description of pathway objects, see the WikiPathways Legend.
Authors
Lobke Meels , Denise Slenter , Eline Sanders , Irene Hemel , Egon Willighagen , Friederike Ehrhart , Eric Weitz , and Finterly HuActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways ONTOX Rare DiseasesAnnotations
Pathway Ontology
glutathione metabolic pathway amino acid metabolic pathway disease pathway glutathione biosynthetic pathway glutathionuria disease pathwayDisease Ontology
hemolytic anemia gamma-glutamyl transpeptidase deficiency autosomal recessive disease metabolic acidosis| Label | Type | Compact URI | Comment |
|---|---|---|---|
| Glutamate | Metabolite | chebi:14321 | |
| Glycine | Metabolite | chebi:15428 | |
| L-gamma-glutamyl amino acid | Metabolite | chebi:15857 | |
| ATP | Metabolite | chebi:15422 | |
| ADP | Metabolite | chebi:16761 | |
| Amino acid | Metabolite | chebi:33704 | |
| Cysteine | Metabolite | chebi:15356 | |
| Glutathione | Metabolite | chebi:16856 | |
| 5-Oxoproline | Metabolite | chebi:16010 | |
| Gamma-glutamylcysteine | Metabolite | chebi:17515 | |
| Cysteinylglycine | Metabolite | chebi:4047 | |
| GGT1 | Protein | uniprot:P19440 | |
| GSS | Protein | uniprot:P48637 | |
| 5-Oxoprolinase | Protein | uniprot:O14841 | |
| GGCT | Protein | uniprot:O75223 | |
| GCLC | Protein | uniprot:P48506 | |
| Dipeptidase | Protein | uniprot:P16444 | DPEP1 |
References
- Glutathionuria: inborn error of metabolism due to tissue deficiency of gamma-glutamyl transpeptidase. Schulman JD, Goodman SI, Mace JW, Patrick AD, Tietze F, Butler EJ. Biochem Biophys Res Commun. 1975 Jul 8;65(1):68–74. PubMed Europe PMC Scholia
- Gamma-glutamylcysteine synthetase deficiency and hemolytic anemia. Beutler E, Moroose R, Kramer L, Gelbart T, Forman L. Blood. 1990 Jan 1;75(1):271–3. PubMed Europe PMC Scholia
- Glutathione synthetase deficiency, an inborn error of metabolism involving the gamma-glutamyl cycle in patients with 5-oxoprolinuria (pyroglutamic aciduria). Wellner VP, Sekura R, Meister A, Larsson A. Proc Natl Acad Sci U S A. 1974 Jun;71(6):2505–9. PubMed Europe PMC Scholia
- The gamma-glutamyl cycle. Diseases associated with specific enzyme deficiencies. Meister A. Ann Intern Med. 1974 Aug;81(2):247–53. PubMed Europe PMC Scholia
- Enzymatic conversion of 5-oxo-L-proline (L-pyrrolidone carboxylate) to L-glutamate coupled with cleavage of adenosine triphosphate to adenosine diphosphate, a reaction in the -glutamyl cycle. Van der Werf P, Orlowski M, Meister A. Proc Natl Acad Sci U S A. 1971 Dec;68(12):2982–5. PubMed Europe PMC Scholia
- 5-oxoprolinuria due to hereditary 5-oxoprolinase deficiency in two brothers--a new inborn error of the gamma-glutamyl cycle. Larsson A, Mattsson B, Wauters EA, van Gool JD, Duran M, Wadman SK. Acta Paediatr Scand. 1981;70(3):301–8. PubMed Europe PMC Scholia
- Prenatal diagnosis of glutathione synthase deficiency. Manning NJ, Davies NP, Olpin SE, Carpenter KH, Smith MF, Pollitt RJ, et al. Prenat Diagn. 1994 Jun;14(6):475–8. PubMed Europe PMC Scholia
- Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Hanigan MH, Ricketts WA. Biochemistry. 1993 Jun 22;32(24):6302–6. PubMed Europe PMC Scholia
- 5-Oxoprolinuria associated with 5-oxoprolinase deficiency; further evidence that this is a benign disorder. Henderson MJ, Larsson A, Carlsson B, Dear PR. J Inherit Metab Dis. 1993;16(6):1051–2. PubMed Europe PMC Scholia
- A pseudo-michaelis quaternary complex in the reverse reaction of a ligase: structure of Escherichia coli B glutathione synthetase complexed with ADP, glutathione, and sulfate at 2.0 A resolution. Hara T, Kato H, Katsube Y, Oda J. Biochemistry. 1996 Sep 17;35(37):11967–74. PubMed Europe PMC Scholia
- The molecular basis of a case of gamma-glutamylcysteine synthetase deficiency. Beutler E, Gelbart T, Kondo T, Matsunaga AT. Blood. 1999 Oct 15;94(8):2890–4. PubMed Europe PMC Scholia
- Long-term clinical outcome in patients with glutathione synthetase deficiency. Ristoff E, Mayatepek E, Larsson A. J Pediatr. 2001 Jul;139(1):79–84. PubMed Europe PMC Scholia
- Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Njålsson R, Ristoff E, Carlsson K, Winkler A, Larsson A, Norgren S. Hum Genet. 2005 Apr;116(5):384–9. PubMed Europe PMC Scholia
- Crystal structures of gamma-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Okada T, Suzuki H, Wada K, Kumagai H, Fukuyama K. Proc Natl Acad Sci U S A. 2006 Apr 25;103(17):6471–6. PubMed Europe PMC Scholia
- The identification and structural characterization of C7orf24 as gamma-glutamyl cyclotransferase. An essential enzyme in the gamma-glutamyl cycle. Oakley AJ, Yamada T, Liu D, Coggan M, Clark AG, Board PG. J Biol Chem. 2008 Aug 8;283(32):22031–42. PubMed Europe PMC Scholia
- γ-glutamyl transpeptidase deficiency caused by a large homozygous intragenic deletion in GGT1. Darin N, Leckström K, Sikora P, Lindgren J, Almén G, Asin-Cayuela J. Eur J Hum Genet. 2018 Jun;26(6):808–17. PubMed Europe PMC Scholia
- Pubmed: 9783642403361