Tyrosine metabolism and related disorders (WP4506)
Homo sapiens

{kind=link}
{kind=link}
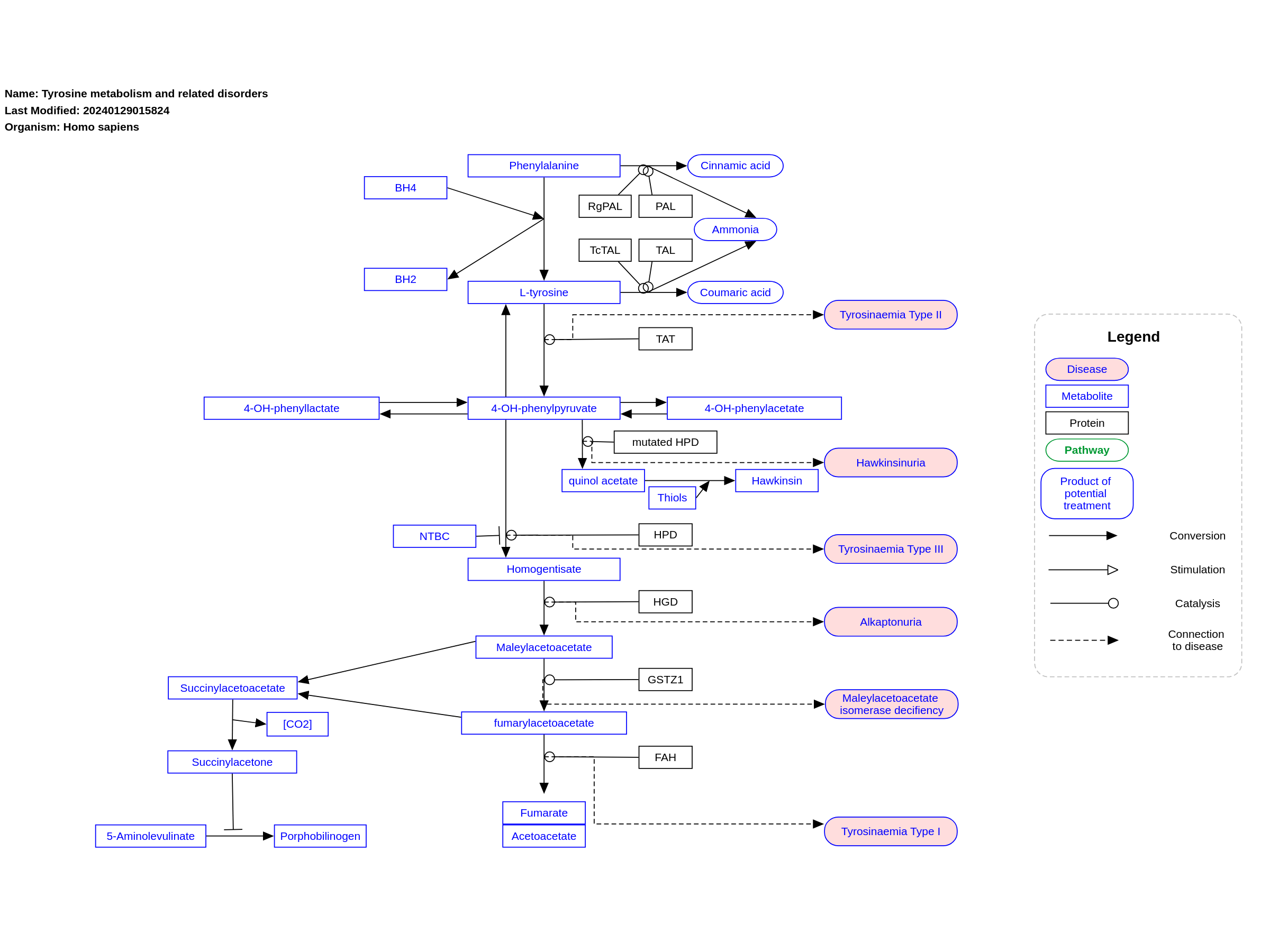
This pathway shows the tyrosine degradation pathway as presented in Edition 5, Chapter 21 of the book of Blau (ISBN 9783030677268); Ed.4 Ch.2. Disorders resulting from an enzyme defect are highlighted in pink. Red frames mark diagnostically important metabolites.
For a description of pathway objects, see the WikiPathways Legend.
Authors
Lauren J. Dupuis , Denise Slenter , Egon Willighagen , Irene Hemel , G. Keulen , Friederike Ehrhart , Agustin Gonzalez-Vicente , Eric Weitz , and Finterly HuActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways Rare DiseasesAnnotations
Pathway Ontology
tyrosine metabolic pathway tyrosine degradation pathway tyrosinemia type I pathway tyrosinemia type II pathway alkaptonuria pathway hawkinsinuria pathway tyrosinemia pathway tyrosinemia type III pathwayDisease Ontology
tyrosinemia type I tyrosinemia type III hawkinsinuria alkaptonuria tyrosinemia type II inherited metabolic disorder| Label | Type | Compact URI | Comment |
|---|---|---|---|
| Cinnamic acid | Metabolite | chebi:27386 | |
| quinol acetate | Metabolite | chebi:31128 | This compound is annotated with an example for quinol acetate (4-hydroxyphenyl acetate). |
| BH4 | Metabolite | chebi:59560 | AKA tetrahydrobiopterin |
| Thiols | Metabolite | chebi:29256 | |
| Phenylalanine | Metabolite | chebi:17295 | AKA L-phenylalanine |
| Ammonia | Metabolite | chebi:16134 | |
| Hawkinsin | Metabolite | pubchem.compound:173909 | aka 2-cystenyl-1,4-dihydroxycyclohexenylacetate |
| Homogentisate | Metabolite | chebi:16169 | |
| [CO2] | Metabolite | chebi:16526 | |
| Succinylacetone | Metabolite | chebi:87897 | |
| Maleylacetoacetate | Metabolite | chebi:17105 | AKA 4-Maleylacetoacetate |
| Fumarate | Metabolite | chebi:29806 | |
| Acetoacetate | Metabolite | chebi:13705 | |
| 4-OH-phenylpyruvate | Metabolite | chebi:36242 | aka 4-Hydroxyphenylpyruvate |
| L-tyrosine | Metabolite | chebi:58315 | |
| NTBC | Metabolite | chebi:50378 | AKA nitisone |
| 4-OH-phenylacetate | Metabolite | chebi:18101 | AKA 4-Hydroxyphenylacetate |
| Succinylacetoacetate | Metabolite | chebi:87999 | |
| fumarylacetoacetate | Metabolite | hmdb:HMDB0062563 | AKA 4-fumarylacetoacetate |
| Porphobilinogen | Metabolite | chebi:17381 | |
| 4-OH-phenyllactate | Metabolite | chebi:36659 | aka p-Hydroxyphenyllactate |
| 5-Aminolevulinate | Metabolite | chebi:17549 | |
| Coumaric acid | Metabolite | chebi:36090 | |
| BH2 | Metabolite | chebi:15642 | AKA dihydrobiopterin |
| GSTZ1 | GeneProduct | uniprot:O43708 | |
| PAL | Protein | eccode:4.3.1.25 | PAL enzymes have side activity towards L-Tyr, mostly from fungi and monocotylic plants. |
| RgPAL | Protein | uniprot:Q2VMT1 | Species: Rhodotorula glutinis |
| mutated HPD | Protein | uniprot:A0A0B4J1R4 | HPD gene, with mutation p.Asn241Ser, leading to a change in function in the protein.Another mutation found to be linked to hawkinsiburia: A heterozygous missense mutation: Ala to Thr change at codon 33 (A33T) [PMID:11073718]AKA 4-hydroxyphenylpyruvate dioxygenase |
| HPD | Protein | uniprot:P32754 | HPD gene, without mutationAKA 4-hydroxyphenylpyruvate dioxygenase |
| FAH | Protein | uniprot:P16930 | AKA Fumarylacetoacetase |
| HGD | Protein | uniprot:Q93099 | AKA Homogentisate 1,2-dioxygenase |
| TAT | Protein | uniprot:P17735 | aka Tyrosine aminotransferase |
| TAL | Protein | eccode:4.3.1.25 | PAL enzymes have side activity towards L-Tyr, mostly from fungi and monocotylic plants. |
| TcTAL | Protein | uniprot:U5TV35 | Species: Trichosporon cutaneum |
References
- Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases [Internet]. Blau N, Duran M, Gibson KM, Dionisi-Vici C. Springer; 2014. 0 p. Available from: https://books.google.com/books/about/Physician_s_Guide_to_the_Diagnosis_Treat.html?hl=&id=wJRBnwEACAAJ OpenLibrary Worldcat
- Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria. Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, et al. Mol Genet Metab. 2000 Nov;71(3):506–10. PubMed Europe PMC Scholia
- Manifestation of hawkinsinuria in a patient compound heterozygous for hawkinsinuria and tyrosinemia III. Item CB, Mihalek I, Lichtarge O, Jalan A, Vodopiutz J, Muhl A, et al. Mol Genet Metab. 2007 Aug;91(4):379–83. PubMed Europe PMC Scholia
- Product analysis and inhibition studies of a causative Asn to Ser variant of 4-hydroxyphenylpyruvate dioxygenase suggest a simple route to the treatment of Hawkinsinuria. Brownlee JM, Heinz B, Bates J, Moran GR. Biochemistry. 2010 Aug 24;49(33):7218–26. PubMed Europe PMC Scholia
- Hypersuccinylacetonaemia and normal liver function in maleylacetoacetate isomerase deficiency. Yang H, Al-Hertani W, Cyr D, Laframboise R, Parizeault G, Wang SP, et al. J Med Genet. 2017 Apr;54(4):241–7. PubMed Europe PMC Scholia
- Exploring the therapeutic potential of modern and ancestral phenylalanine/tyrosine ammonia-lyases as supplementary treatment of hereditary tyrosinemia. Hendrikse NM, Holmberg Larsson A, Svensson Gelius S, Kuprin S, Nordling E, Syrén PO. Sci Rep. 2020 Jan 28;10(1):1315. PubMed Europe PMC Scholia